A Rare Case of Skull Base Phosphaturic Mesenchymal Tumor
Affiliations
- 1 Medicover Cancer Institute, Hyderabad, India
Phosphaturic mesenchymal tumor (PMT) is a rare type of tumor that presents as a paraneoplastic syndrome causing tumor-induced osteomalacia. So far, close to 500 cases have been reported in the literature, making it a rare entity in clinical practice. The most common sites of PMT involvement are extremities. Here, we report a rare case of PMT involving the skull base.
Case Summary
A 55-year-old woman presented with a history of multiple bone fractures, headache, double vision, left-ear impaired hearing, and impaired urinary control. In 2018, she sustained a left hip fracture, in 2019 a right hip fracture, in 2020 a right ulnar fracture following a fall, and in 2021 a left ulnar fracture. The patient was evaluated by an orthopedic surgeon initially and later by an endocrinologist. Imaging with PET/CT and MRI in January 2022 showed features suggestive of a large lytic lesion involving the skull base. PET/CT revealed an intensely avid lytic lesion with an enhancing soft-tissue component involving the left petrous temporal bone extending into the squamous temporal, occipital bone, sphenoid bone, arch of atlas and clivus. MRI showed extensive hypointense and heterogeneously hyperintense lesions at the left petrous temporal bone, mastoid and part of the occipital bone, with extensive destruction of the bone, which was replaced by soft-tissue component lesions and cystic areas. There was extensive destruction of bone at the skull base involving the clivus and basi-sphenoid.
Biopsy (January 2022) and immunohistochemistry (IHC) findings (S100 positive, DOG1 noncontributory, and Ki67 1%-2%) were suggestive of a phosphaturic mesenchymal tumor. Serum FGF-23 (fibroblast growth factor) levels were 2088 pg/mL (biological reference range, 23.2-95.4 pg/mL), and serum phosphorus levels were 1.6 mg/dL (biological reference range, 2.5-4.5 mg/dL).
The case was discussed in our tumor board meeting with the decision to consider surgical debulking followed by radiation therapy as R0 resection (surgical resection with negative margins) was considered not feasible in view of extensive skull base involvement. The patient refused to undergo surgery after discussing potential benefits and complexities of the procedure with the neurosurgeon, and instead opted for external-beam radiation therapy.
Treatment Details
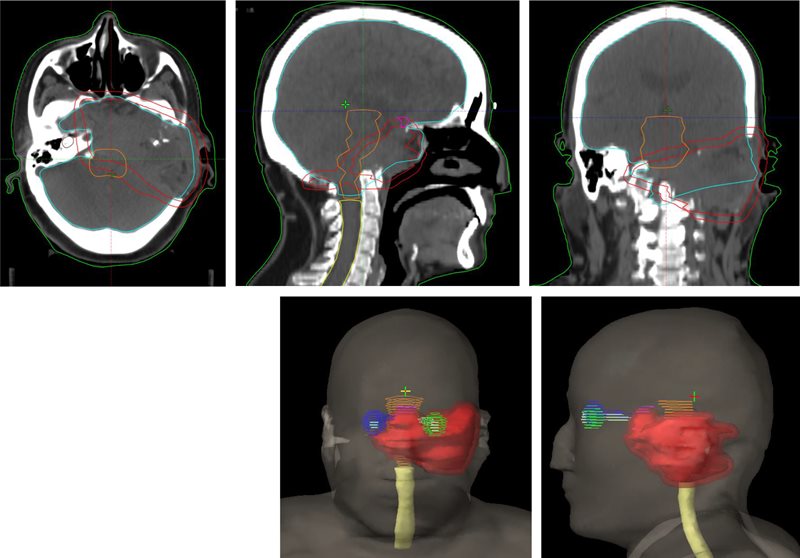
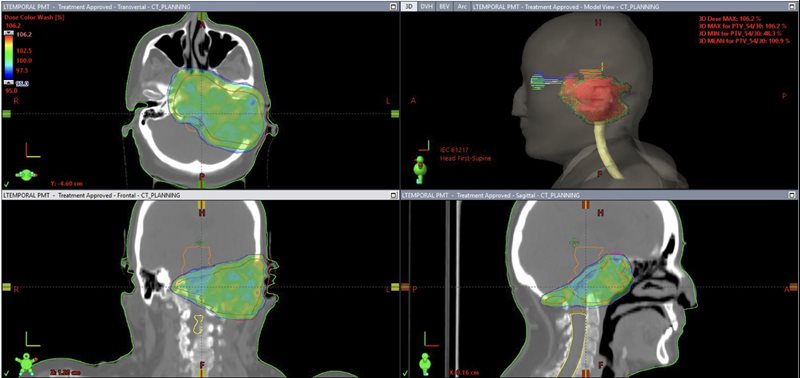
CT simulation was performed on a 16-slice PET/CT simulator. The patient was immobilized in the treatment position using a thermoplastic mask. CT images were acquired in 1.25-mm slice thickness from the vertex of the skull to the mid-chest. PET/CT image fusion was performed and target volumes and organs at risk were contoured on the planning CT scan ( Figure 1 ). The planning target volume (PTV) included the gross tumor volume with a uniform 0.5-cm margin for setup errors. Treatment planning was performed using the Varian Eclipse treatment planning system. Prescription dose was 54 Gy in 30 fractions, 1.8 Gy per fraction to the PTV ( Figure 2 ). Treatment was delivered with the Varian TrueBeam linear accelerator using 6 MV photons with the Varian RapidArc technique. The patient tolerated the treatment well. Acute radiation side effects such as nausea, headache, and skin hyperpigmentation were managed conservatively with symptomatic medications such as oral ondansetron and paracetamol. Follow-up at 6 months revealed no symptom progression and mild improvement regarding headaches. The patient experienced no bone fractures after the treatment.
Planning CT images with contours of lesion (red: planning target volume [PTV]) and adjacent normal structures (green: left retina; dark blue: right retina; cyan: brain; orange: brainstem; yellow: spinal cord).
Treatment plan showing radiation dose distribution (green color) in color wash and planning target volume (PTV) (red contour).

Imaging Findings
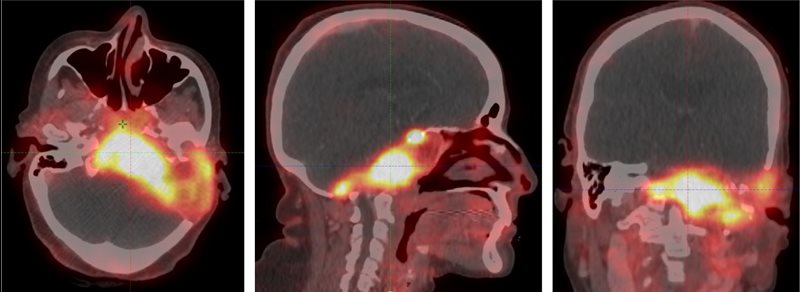
A gallium 68 dotatate PET/CT ( Figure 3 ) performed in January 2022 revealed an intensely avid lytic lesion with an enhancing soft-tissue component involving the left petrous temporal bone extending into the squamous temporal, occipital bone, sphenoid bone, arch of atlas, and clivus, with a standardized uptake value (SUV) max of 10. Anteriorly, it was involving the longus coli muscles, extending into the nasopharynx and sphenoid sinus. Inferiorly, it was abutting the posterior margin of the left parotid gland. Superomedially, it was extending up to the cavernous sinus, encasing the left petrous internal carotid artery and compressing the left internal jugular vein. It was causing a mass effect on the left cerebellar hemisphere, brainstem, and fourth ventricle. Multiple bony fractures were noticed in the skeleton. No distant metastasis was detected.
Gallium 68 dotatate PET/CT images showing a tracer avid lytic lesion at the skull base.
MRI of the brain ( Figure 4 ) performed in January 2022 showed extensive T1 iso- to hypointense and T2 heterogeneously hyperintense lesions at the left petrous temporal bone, mastoid, part of the occipital bone with extensive destruction of the bone, and replaced with soft-tissue component lesions and cystic areas. There was extensive destruction of the bone at the skull base involving the clivus and basi-sphenoid. The lesion was extending through the floor of the pituitary fossa into the sella. There was destruction of the clinoid process on both sides and greater wing of the sphenoid on the left side. Inferior extension of the lesion was causing bony destruction of the left occipital condyle and left half of the anterior arch of C1 vertebra. A postcontrast study showed extensive and homogeneous enhancement of the lesion, measuring approximately 6.6 × 5.5 × 3.4 cm. The left internal auditory canal and VII/VIII cranial nerves were not visualized separately. MR spectroscopy revealed a high possibility of a malignant lesion with elevated choline and an altered choline/N-acetylaspartate ratio.
MRIs showing the lesion at the skull base: T1 with contrast (A-C),T1 plain (D), T2 (E), fluid-attenuated inversion recovery (FLAIR) (F), and diffusion-weighted imaging (DWI) (G).

Diagnosis
The patient was diagnosed with PMT, which often tends to be small and can be located anywhere in the body, mimicking many other common tumors of the bone and soft tissue. Differential diagnosis includes chondromyxoid fibroma, chondroblastoma, aneurysmal bone cyst, glomus jugulare, chordoma, vestibular schwannoma, and osteosarcoma.
Discussion
PMT is a rare entity that usually presents with a clinical picture of tumor-induced osteomalacia (TIO).1 - 3 To date, fewer than 500 cases have been reported in the literature.4 Osteomalacia is a metabolic disorder in which there is insufficient mineralization of the mature bone. The most common cause of osteomalacia is vitamin D deficiency. Other rare causes include inborn errors of metabolism and chronic kidney disease. TIO is a type of paraneoplastic syndrome that can be seen in osteoblastoma, osteosarcoma, hemangiopericytoma, and plasmacytoma.4
TIO was first reported by McCance in a 15-year-old teenager who presented with weakness and gait disturbance, along with hypophosphatemia.4 The term phosphaturic mesenchymal tumor was coined by Wiedner and Santa Cruz in 1987.5 PMT was included in the WHO 2013 classification of tumors for bone and soft tissue.6
The pathophysiology of TIO involves excessive production of FGF-23 by the tumor cells, which is a type of phosphate-regulating substance in the body.4 FGF-23 reduces phosphate reabsorption in the proximal renal tubules, leading to excessive renal excretion of phosphates. FGF-23 also increases bone resorption of calcium and phosphate, and decreases intestinal absorption of calcium and phosphate, decreasing bone mineralization.4
PMT is characteristically a benign tumor.5, 6 However, local recurrences and malignant behavior in the form of lung metastasis have also been reported in the literature.5, 7 Hence, patients need to be followed up with appropriate imaging of the chest, serum phosphates, and FGF-23 levels to detect any recurrence.3, 6, 8, 9
PMT commonly affects the extremities of middle-aged patients and can originate from the bone or soft tissue. It often tends to be small and can be located anywhere in the body, mimicking many other common tumors of the bone and soft tissue.10
The diagnosis is often delayed due to vague symptomatology and low degree of suspicion of PMT.8 Patients usually present with recurrent fractures, bone pains, muscle pains, and generalized weakness.9 Biochemical findings include hypophosphatemia, hyperphosphaturia, and elevated levels of FGF-23.4
Imaging modalities for diagnosis may include CT, MRI, FDG PET/CT, dotatate PET/CT, and a Tc-99m sestamibi scan. Radiographic features of PMTs have been described recently in detail.11 On CT scans, bone lesions are typically osteolytic, show a narrow zone of transition, and contain internal matrix.12 On MRI, they are usually T1 isointense, T2 hyperintense, and solidly enhancing, with areas of dark T2 signal.12
Histologically, there are 4 types of PMTs: osteoblastoma-like variant, nonossifying fibroma-like variant, ossifying fibroma-like variant, and mixed connective tissue variant.1, 6 The most common type is the mixed connective tissue variant.4 Morphological differential diagnosis includes chondromyxoid fibroma, chondroblastoma, aneurysmal bone cyst, glomus jugulare, chordoma, vestibular schwannoma, and osteosarcoma.4 Histological findings of benign cartilage-forming lesion in correlation with IHC, typical clinical presentation of recurrent fractures, and biochemical profile showing hypophosphatemia with elevated levels of FGF-23 confirm the diagnosis of PMT and differentiate it from other differentials.
Treatment options include surgery for resectable lesions. Complete surgical resection with negative surgical margins of at least 10 mm is the mainstay treatment.4 Radiation therapy is used for surgically or medically inoperable patients, incompletely resected tumors, or positive margins after resection.4 Although the detailed mechanism of the effectiveness of radiation therapy for PMT is unclear, the obstruction and fibrosis of the tumor vessels could occur, thus inhibiting growth, similar to the mechanism observed in other hormone- or cytokine-producing tumors.13, 14 It is important to use a high-precision radiation therapy technique such as intensity-modulated radiation therapy or volumetric-modulated arc therapy for critical sites such as the skull base or the head and neck region to spare the adjacent normal anatomical structures. Data regarding radiation therapy doses for PMT are very limited. As discussed in the case report by Shah et al, 54 Gy in 30 fractions was used in the postoperative adjuvant setting.15 Another case report by Uramoto et al mentioned the use of 66 Gy in 33 fractions after marginal resection of PMT of the tongue.16 In addition to local control, radiation therapy was found effective in improving oncogenic osteomalacia.17 Radiofrequency ablation (RFA) can be used for small bony lesions.4 Concomitant medical management includes phosphorus and calcitriol supplementation.3, 6
Conclusion
PMT is a rare histological type of mesenchymal tumor involving the bone and soft tissue. In most cases, it is benign, but a few cases of malignant PMTs have also been reported. PMT is one of the most common causes of TIO. Clinical presentation of recurrent fractures, osteomalacia with biochemical findings of hypophosphatemia, and elevated levels of FGF-23 suggest a possible diagnosis of PMT. Surgery with negative margins is the mainstay treatment for operable lesions. Other nonsurgical treatment options for inoperable or incompletely resected tumors include radiation therapy and RFA. Here, we report a case of PMT of the skull base that was considered inoperable and was treated with definitive radiation therapy.
References
Citation
Mirza Athar Ali, MD, Muntimadugu Babaiah, MD, Prabhakar Mariappan, MD, Saadvik Raghuram, DM, Krishna Reddy Thaduri, DM, Muthulingam Shunmugavel, MSc, Soundarya YSB, MSc, Oviya Manohar, MSc, Surya Simhareddy Molakala, MBBS. A Rare Case of Skull Base Phosphaturic Mesenchymal Tumor. Appl Radiat Oncol. 2024;(3):48 - 53.
doi:10.37549/ARO-D-24-00004
September 1, 2024