Proton therapy for radiation-induced parameningeal rhabdomyosarcoma
Images
Retinoblastoma is the most common childhood primary intraocular tumor, and occurs primarily in young children with 95% of cases arising before age 5. The RB1 gene on chromosome 13q14 was the first tumor suppressor gene discovered, and inactivation of both alleles of the RB1 gene is the initiating event in the formation of retinoblastoma.1,2 Patients with heritable retinoblastoma have a constitutional RB1 mutation and frequently develop bilateral retinoblastoma. One-fourth to one-third of patients with retinoblastoma present with bilateral disease and all of these cases are heritable. In addition, about 13% of cases of unilateral retinoblastoma are heritable.2-4 However, the majority of heritable cases have a de novo germline mutation with no family history of retinoblastoma.2
The incidence of retinoblastoma has remained constant worldwide at 1 case per 16,000 to 18,000 live births.5,6 This corresponds to about 8,000 new cases annually, mostly concentrated in Asia and Africa where there are large populations with high birth rates. Great strides have been made in treating retinoblastoma in the developed world with 3% to 5% mortality in the United States, Canada, and Europe; however, mortality remains high at 40% to 70% in Asia and Africa.2 Radiotherapy has a long-standing and well-established role in the adjuvant treatment of children with retinoblastoma at a high risk of local progression. However, the exquisite radiosensitivity of normal tissue in very young children and the increased incidence of radiation carcinogenesis in patients with RB1 mutations have prompted new approaches of radiation avoidance or newer technology that might reduce the risk of collateral radiation injury in retinoblastoma patients. This case illustrates a unique example of a radiation-induced rhabdomyosarcoma in a child previously radiated for retinoblastoma in which proton therapy was recommended in hopes of mitigating additional radiation side effects.
Pediatric case
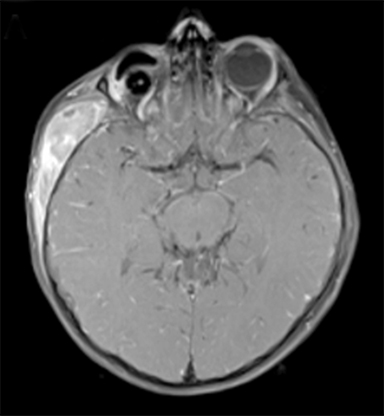
At 5 months, our patient, a white male, was diagnosed with bilateral retinoblastoma. His right eye was staged as Group D, according to the International Classification for Intraocular Retinoblastoma (large tumor with associated retinal detachment), and his left eye was staged as Group B (3 small peripheral tumors). The decision was made to treat with carboplatin, vincristine, and etoposide (CVE) chemotherapy in view of bilateral disease rather than proceeding with immediate enucleation. After an anaphylactic reaction to the first carboplatin infusion, he received ifosfamide, etoposide, and vincristine (IVE) for 6 cycles. His initial response to the chemotherapy was good with a response at all tumor sites. Unfortunately, at the end of the treatment examination he was noted to have an extensive relapse (local and vitreous base) in the right eye and a relapse near the macula in the left eye. In view of the early relapse, the prior use of ifosfamide (a major component of any chemotherapy relapse strategy) and the prospect of no useful vision in the right eye, he underwent enucleation of the right eye and radiotherapy to 40 Gy to the left eye. A left lateral lens-sparing 6-MV technique exiting through the empty right socket was used, prescribed to 2.5-cm depth for 20 fractions for 4 weeks using a vacuum fixation of the eye (Figure 1). Following completion of this treatment, the patient has had no evidence of recurrence of his retinoblastoma. There was no family history of retinoblastoma, although on examination his father was noted to have a retinal scar (retinoma or regressed retinoblastoma). The family declined genetic testing.
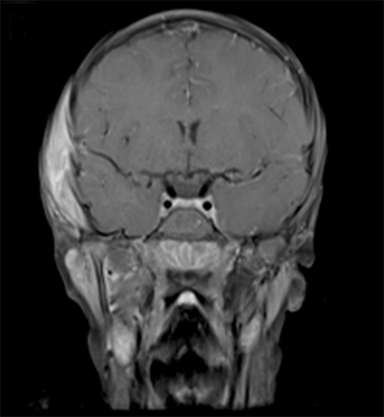
At 6 years old, now with an additional diagnosis of autistic spectrum disorder, our patient re-presented to his general practitioner with a 1-week history of painless swelling of the right side of his face. With no prior history of trauma, an infectious cause was presumed and he received a course of amoxicillin clavulanate. When the swelling did not improve after 48 hours on the antibiotics, he was referred to his local ophthalmologist and then to the retinoblastoma specialist for further assessment. On examination, he was found to have a 4-×-4-cm diffuse swelling over the right zygomatic region with no palpable adenopathy. Ultrasound of the right face revealed an oval-shaped heterogeneously hypoechoic vascularized lesion that measured 4.5-×-1.1 cm overlying the right zygomatic bone with no disruption of the underlying bone seen. Magnetic resonance imaging (MRI) of the face and neck revealed a well-defined solid enhancing mass within the right infratemporal fossa with restricted diffusion in keeping with tumor. There was overlying soft tissue edema that extended anteriorly into the muscles of mastication and buccal fat on the right. There were some enlarged right parotid and right upper cervical chain lymph nodes (Figure 2). The lesion was noted to involve the right zygomatic arch, right skull base, and mandibular ramus.
Biopsy was performed of the mass overlying the right zygomatic bone as well as the right parotid lymph node and the right upper cervical lymph node. All 3 specimens showed rhabdomyosarcoma and favored alveolar type because of strong myogenin staining and cell morphology despite absence of PAX3-FOXO1 and PAX7-FOXO1 fusion transcripts by RT-PCR.
Approximately 6 weeks after presenting, the known right temporal mass had enlarged and now measured 5.7-×-2.8 cm. The additional right parotid lesion measured 2-×-1.3 cm. The lesion in the right submandibular region measured 3.6-×-2 cm. There were significantly enlarged right cervical nodes with smaller nodes present in the left side of the neck.
Metastatic workup was negative and the patient was staged as T2bN1M0, Intergroup Rhabdomyosarcoma Study (IRS) stage 3, group III. The decision was made to treat the patient according to the very high-risk group (H) of the European Pediatric Soft Tissue Sarcoma Study Group (EpSSG) RMS 2005 study (the patient was technically eligible for enrollment because this was a second tumor) with the application of proton therapy for local control. He commenced chemotherapy with ifosfamide, vincristine, actinomycin-D, and doxorubicin (IVADo) 2 months after presenting with right facial swelling. He tolerated chemotherapy poorly and lost 4 kg in the first 2 weeks of treatment. A percutaneous endoscopic gastrostomy (PEG) tube was placed to maintain his nutrition. He had significant nausea, vomiting, mucositis, and anorexia and was hospitalized through the majority of his first 4 cycles of chemotherapy. MRI after completion of 4 cycles of IVADo demonstrated an almost complete resolution of the right temporal mass and significant improvement in the right cervical adenopathy.
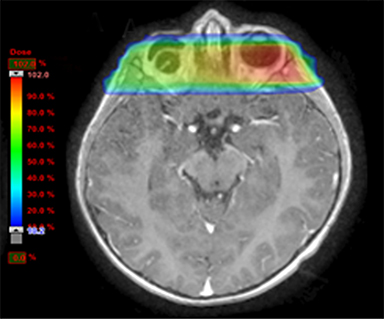
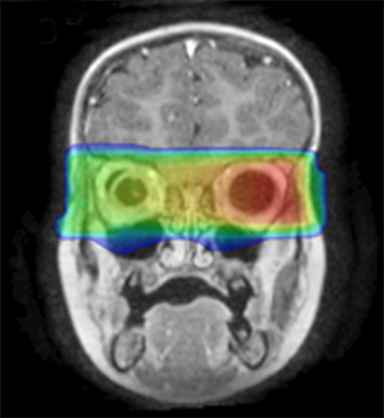
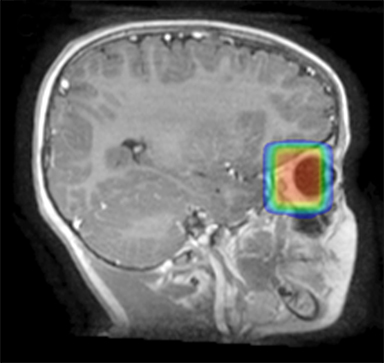
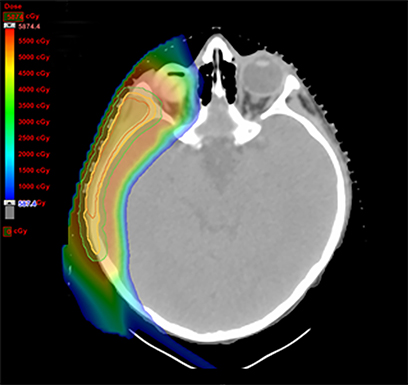
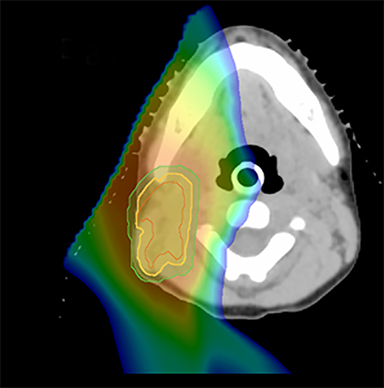
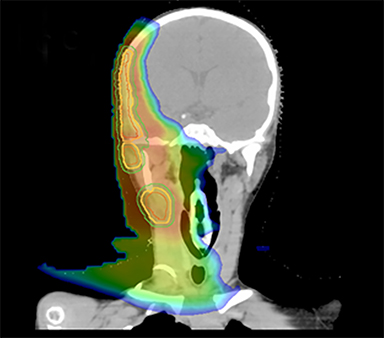
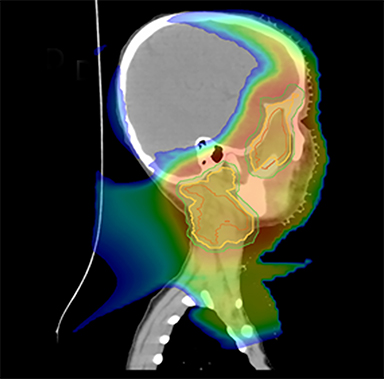
As per protocol, he continued on ifosfamide, vincristine, and actinomycin D (IVA) maintenance for 2 cycles prior to starting radiotherapy. MRI after the second cycle of IVA chemotherapy demonstrated no remaining disease. He commenced proton radiotherapy in conjunction with the third cycle of IVA chemotherapy. A total of 41.4 Gy (relative biological effectiveness [RBE]) was delivered at 1.8 Gy (RBE) per daily fraction (23 fractions) to a target volume based on the pre-chemotherapy extent of disease, including the right cervical nodal chain, and the right parotid region with a 2-field 3-dimensional conformal passive double-scattering proton plan. An additional 9 Gy (RBE) at 1.8 Gy (RBE) per daily fraction (5 fractions) was given to a target volume based on the post-chemotherapy volume for a total dose of 50.4 Gy (RBE) in 28 daily fractions (Figure 3).
The patient completed adjuvant chemotherapy with 6 cycles total of IVA, and then received maintenance therapy with vinorelbine and oral cyclophosphamide for 6 cycles. Currently he is alive and well 12 months after completing treatment. He has normal endocrine function, no abnormal audiology, no additional eye changes beyond his original diagnosis, and a reduced fractional shortening (28%) secondary to his anthracycline exposure. He continues experiencing nutritional difficulties and relies on his percutaneous endoscopic gastrostomy (PEG) tube for feeding, and his educational progress and difficulties pertaining to his autistic spectrum disorder have been compounded by the social complexities of his recent treatment away from his home.
Discussion
Even with successful treatment of retinoblastoma, patients with a constitutional RB1 mutation are highly susceptible to developing secondary malignant neoplasms (SMNs). The risk of SMNs in patients with heritable retinoblastoma is estimated to be 36% to 48% at 50 years.7,8 This translates to an increased mortality from SMNs at 50 years after diagnosis in patients with heritable retinoblastoma of 17% to 26%.9,10 Recently, patients with an inherited germline mutation were found to have an increased risk for SMN compared to those with a de novo germline mutation, largely due to an increased risk for melanoma, and there was no increased risk for bone or soft tissue sarcoma.11 The increased risk of SMN may have a genetic basis, as patients with recurrent nonsense mutations have been found to have an increased risk of SMNs and those with low-penetrance mutations have a lower risk.12
Radiotherapy results in a threefold increased risk of SMNs in patients with heritable retinoblastoma. An analysis of the Dutch retinoblastoma registry found that the risk of SMN was 13.3% at 40 years in patients with heritable retinoblastoma who did not receive radiotherapy, and this increased to 33.2% in those who received radiotherapy.13 A cohort of heritable retinoblastoma patients from the United States demonstrated a cumulative risk of SMN of 21% at 50 years in those who did not receive radiotherapy, compared to 38% in those who received radiotherapy.8 Soft tissue sarcomas account for a sizeable proportion of these SMNs in patients with heritable retinoblastoma who receive radiotherapy, with a 13% cumulative incidence at 50 years.14 A further analysis demonstrated that irradiated survivors had an increased risk of death from SMNs with a standard mortality ratio (SMR) of 3 times that of nonirradiated survivors. Patients irradiated at 12 months or younger had a further increased risk of death from SMNs with an SMR of 2 compared to those irradiated older than 12 months.10
Of the heritable retinoblastoma patients who receive radiotherapy and develop an SMN, 40% to 70% of these will occur in the radiation field.7,13,15 SMNs that occur in a radiation field develop at an earlier age than those that occur outside of a radiation field or those in patients who did not receive radiation therapy. One review of over 600 retinoblastoma patients with SMNs found a median age of diagnosis of 9 years for tumors in the radiation field. Approximately 70% of these tumors will be bone or soft tissue sarcomas. There also appears to be a radiation dose response for developing an in-field sarcoma, with a risk threshold as low as 5 Gy.16 Patients with rhabdomyosarcoma had the youngest age of onset with a median age of 7 years,13 very similar to our patient.
Radiation technique may also modify the risk of SMNs in patients with heritable retinoblastoma. One analysis found the cumulative risk of SMNs was 32.9% at 40 years in patients with heritable retinoblastoma treated with orthovoltage (prior to 1960), and this decreased to 26.3% in those treated with techniques that generated less scatter.7 Conformal photon techniques such as volumetric arc therapy may provide better conformality and may better spare the orbital bone and brain from higher doses compared to 2- or 3-dimensional conformal radiotherapy. However, these techniques also generate a higher integral dose of tissue receiving >5 Gy, which may increase the risk SMNs in patients with a germline mutation.17-19 Dosimetric analyses have demonstrated that proton therapy results in optimal target coverage with the lowest dose to the surrounding orbital bone and reduced integral dose.18,19 Proton therapy may reduce the risk of SMN in patients with germline mutations compared to electron or photon techniques. The first report on SMN of patients with retinoblastoma treated with proton therapy demonstrated a 10-year incidence of only 5%. Of the 52 patients from that single institution series, 85% had heritable retinoblastoma and the 1 patient with SMN suffered from a femoral osteosarcoma.20 While promising, this small series with a median 6.9 years of follow-up will need continued follow-up to confirm these results.
The in-field alveolar rhabdomyosarcoma with lymph node metastases that developed in our patient occurred in a typical time frame from his initial radiation for his retinoblastoma. Patients with clinical group III (unresectable at diagnosis) rhabdomyosarcoma who were treated on IRS IV had a 5-year freedom from treatment failure rate of 77%; however, this was lower at 63% for those with parameningeal primaries.21 Patients on IRS IV with lymph node metastases had a significantly worse 5-year freedom from treatment failure rate at 46% compared to 73% for patients with N0 disease. This difference was more pronounced in those with alveolar histology,22 although the adverse prognosis seen with alveolar histology is likely driven by the 70% to 80% of these patients who have a PAX-FOXO1 fusion gene. A recent analysis attempted to combine stage, age and molecular data to develop a better risk stratification for patients with rhabdomyosarcoma and defined 4 clinicomolecular risk groups. Patients such as ours who were stage 3 and did not have a PAX7-FOXO1 or PAX3-FOXO1 fusion were placed in clinicomolecular risk group 2 with a 5-year overall survival rate of 65%.23
Our patient received standard European chemotherapy and the decision was made to use proton radiotherapy for local control because it has been shown to reduce the dose to the optic structures, brain, hypothalamus, pituitary, and all contralateral structures in a parameningeal rhabdomyosarcoma.24 The reduced integral dose was also thought to be particularly important in this patient with a history of heritable retinoblastoma and already 1 radiation-induced SMN. The first clinical series of patients with parameningeal rhabdomyosarcoma treated with proton radiotherapy demonstrated comparable tumor control, with survival and toxicity comparing favorably to contemporary series.25 In our patient, the proton plan delivered no dose to his optic nerve, pituitary, hypothalamus, or contralateral facial structures. Less than 50% of his ipsilateral temporal lobe received >20 Gy. His bilateral hippocampi and contralateral (left) temporal lobe were entirely spared, which was critical given his pre-existing developmental delay and autistic spectrum disorder.
Patients with heritable retinoblastoma who survive an SMN remain at risk for further neoplasms. Examination of the Dutch registry demonstrated a 7-fold hazard ratio for the risk of a third malignancy compared to the risk of a second malignancy. All of the third malignancies developed within 20 years of the SMN and only one of 11 were in the radiation field. Developing a third malignancy was also associated with a fivefold worse survival than developing a second malignancy.26 Of 211 patients with SMN after retinoblastoma followed at a clinic in New York, the risk of developing a third primary was 22% at 10 years, and the 10-year survival rate for patients with a third malignancy was 30%. The median time to developing a third malignancy was 6 years.27
Patients with heritable retinoblastoma are at an increased risk of SMN, and this risk is significantly increased by radiotherapy. Using proton therapy over photon therapy may reduce this risk as well as other late effects of radiotherapy. In this unique setting where radiotherapy is determined to be necessary for an optimal chance of disease control, then proton therapy specifically should be considered to minimize long-term toxicity and the risk of subsequent malignancies.
References
- Friend SH, Bernards R, Rogelj S, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323:643-646.
- Dimaras H, Kimani K, Dimba EA, et al. Retinoblastoma. Lancet. 2012;379:1436-1446.
- Broaddus E, Topham A, Singh AD. Incidence of retinoblastoma in the USA: 1975-2004. Br J Ophthalmol. 2009;93:21-23.
- Mallipatna AC, Sutherland JE, Gallie BL, et al. Management and outcome of unilateral retinoblastoma. J AAPOS. 2009;13:546-550.
- Seregard S, Lundell G, Svedberg H, et al. Incidence of retinoblastoma from 1958 to 1998 in Northern Europe: Advantages of birth cohort analysis. Ophthalmol. 2004;111:1228-1232.
- Kivela T. The epidemiological challenge of the most frequent eye cancer: Retinoblastoma, an issue of birth and death. Br J Ophthalmol. 2009;93: 1129-1131.
- Kleinerman RA, Tucker MA, Tarone RE, et al. Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma: An extended follow-up. J Clin Oncol. 2005;23:2272-2279.
- MacCarthy A, Bayne AM, Draper GJ, et al. Non-ocular tumours following retinoblastoma in Great Britain 1951 to 2004. Br J Ophthalmol. 2009;93: 1159-1162.
- Marees T, van Leeuwen FE, de Boer MR, et al. Cancer mortality in long-term survivors of retinoblastoma. Eur J Cancer. 2009;45:3245-3253.
- Yu CL, Tucker MA, Abramson DH, et al. Cause-specific mortality in long-term survivors of retinoblastoma. J Natl Cancer Inst. 2009;101:581-591.
- Kleinerman RA, Yu CL, Little MP, et al. Variation of second cancer risk by family history of retinoblastoma among long-term survivors. J Clin Oncol. 2012;30:950-957.
- Dommering CJ, Marees T, van der Hout AH, et al. RB1 mutations and second primary malignancies after hereditary retinoblastoma. Fam Cancer. 2012;11:225-233.
- Woo KI, Harbour JW. Review of 676 second primary tumors in patients with retinoblastoma: Association between age at onset and tumor type. Arch Ophthalmol. 2010;128:865-870.
- Kleinerman RA, Tucker MA, Abramson DH, et al. Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J Natl Cancer Inst. 2007;99:24-31.
- Marees T, Moll AC, Imhof SM, et al. Risk of second malignancies in survivors of retinoblastoma: More than 40 years of follow-up. J Natl Cancer Inst. 2008;100:1771-1779.
- Wong FL, Boice JD, Jr., Abramson DH, et al. Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk. JAMA. 1997;278: 1262-1267.
- Eldebawy E, Parker W, Abdel Rahman W, et al. Dosimetric study of current treatment options for radiotherapy in retinoblastoma. Int J Radiat Oncol Biol Phys. 2012;82:e501-505.
- Lee CT, Bilton SD, Famiglietti RM, et al. Treatment planning with protons for pediatric retinoblastoma, medulloblastoma, and pelvic sarcoma: How do protons compare with other conformal techniques? Int J Radiat Oncol Biol Phys. 2005;63:362-372.
- Krengli M, Hug EB, Adams JA, et al. Proton radiation therapy for retinoblastoma: Comparison of various intraocular tumor locations and beam arrangements. Int J Radiat Oncol Biol Phys. 2005;61:583-593.
- Sethi RV, Shih HA, Yeap BY, et al. Second nonocular tumors among survivors of retinoblastoma treated with contemporary photon and proton radiotherapy. Cancer. 2014;120:126-133.
- Rodeberg DA, Stoner JA, Hayes-Jordan A, et al. Prognostic significance of tumor response at the end of therapy in group III rhabdomyosarcoma: A report from the children’s oncology group. J Clin Oncol. 2009;27:3705-3711.
- Rodeberg DA, Garcia-Henriquez N, Lyden ER, et al. Prognostic significance and tumor biology of regional lymph node disease in patients with rhabdomyosarcoma: A report from the Children’s Oncology Group. J Clin Oncol. 2011;29:1304-1311.
- Missiaglia E, Williamson D, Chisholm J, et al. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J Clin Oncol. 2012;30:1670-1677.
- Kozak KR, Adams J, Krejcarek SJ, et al. A dosimetric comparison of proton and intensity-modulated photon radiotherapy for pediatric parameningeal rhabdomyosarcomas. Int J Radiat Oncol Biol Phys. 2009;74:179-186.
- Childs SK, Kozak KR, Friedmann AM, et al. Proton radiotherapy for parameningeal rhabdomyosarcoma: Clinical outcomes and late effects. Int J Radiat Oncol Biol Phys. 2012;82: 635-642.
- Marees T, van Leeuwen FE, Schaapveld M, et al. Risk of third malignancies and death after a second malignancy in retinoblastoma survivors. Eur J Cancer. 2010;46:2052-2058.
- Abramson DH, Melson MR, Dunkel IJ, et al. Third (fourth and fifth) nonocular tumors in survivors of retinoblastoma. Ophthalmol. 2001;108:1868-1876.
Latest Review Articles
- A Narrative Review on Radiation Oncology Physician Well-Being in the United States
- A Novel Framework to Define and Prognosticate Visual Outcomes Following Fractionated Radiation Therapy for Optic Nerve Sheath
- A Practical Method to Prolong Expiratory Breath Holds for Abdominal Stereotactic Body Radiation Therapy
- Radiation Therapy-Induced Toxicity in a Breast Cancer Patient With Variance of Unknown Significance in the Ataxia Telangiectasia
- Late Effects of Pelvic Radiation Therapy in the Female Patient: A Comprehensive Review